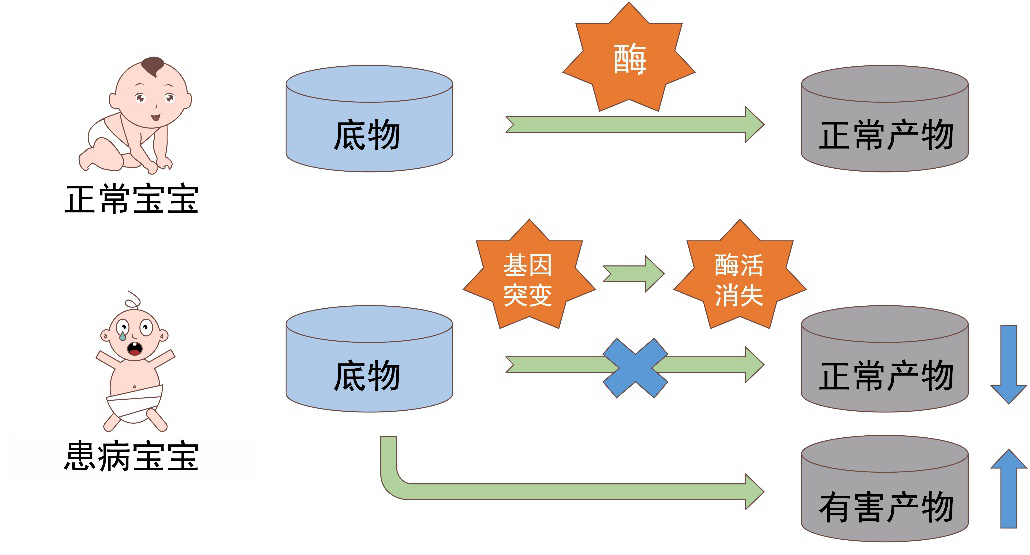
高危代谢病新生儿酶学筛查
什么是溶酶体病
溶酶体是人体的一个细胞器,内含有60多种酸性水解酶,这些酶可以将各种各样的大分子垃圾降解为小分子,从而被排出体外,因而被称为人体的 “垃圾处理器”。如果溶酶体内的任一种酶发生了异常,体内的大分子“垃圾”不能被降解而在细胞内外堆积,就会形成溶酶体病。法布里病,戈谢病、庞贝氏病及粘多糖贮积症I型是常四种常见的溶酶体病。
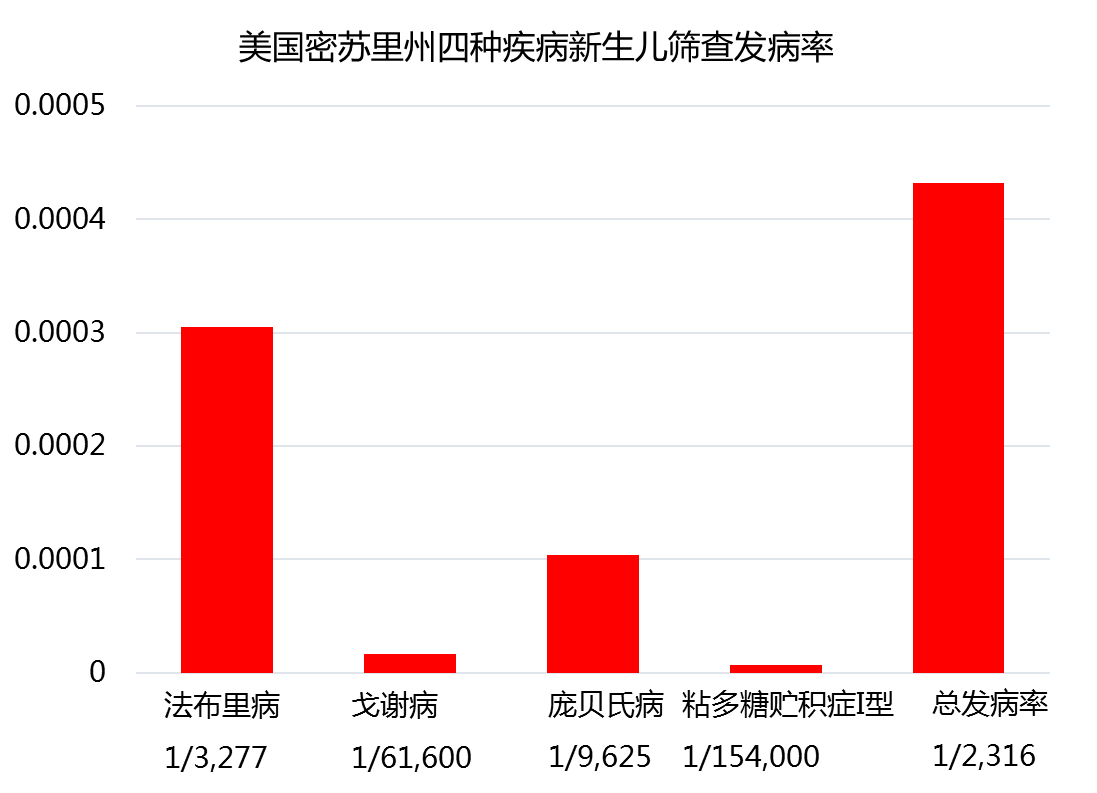
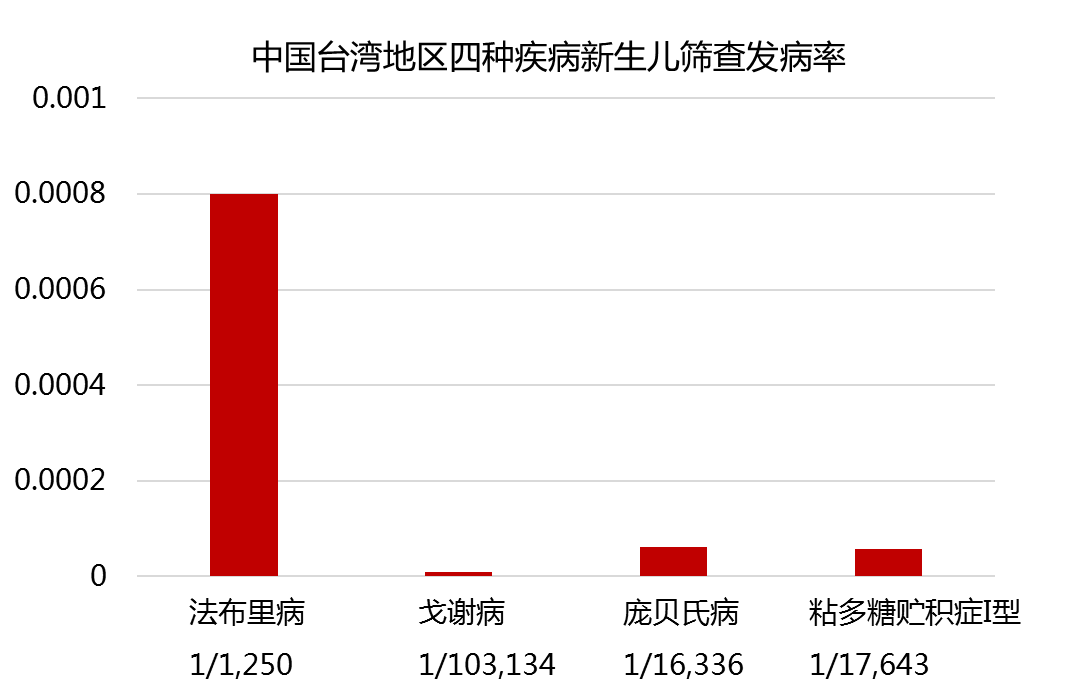
新生儿筛查发病率
许多国家和地区已经开展了这四种疾病的新生儿筛查,中国台湾地区在2009年已经把多项溶酶体病列入了新生儿筛查项目。文献报道美国密苏里州对2013年11月到2017年10月的308000例新生儿进行了这四种疾病的筛查,共确诊133名新生儿患有四种疾病中的1种,其总的发病率约为1/2000。
中国台湾地区庞贝氏病筛查的情况
2019年2月第15届世界溶酶体大会,中国台湾地区介绍早期治疗对于早发性庞贝氏病的作用,并呼吁全世界应该普及庞贝氏病的新生儿筛查。
中国台湾地区筛查情况介绍:台北荣民总医院从2008年开始针对庞贝氏病新生儿筛查,共测试了多达三分之二的台湾新生儿。从2010年开始,他们简化了流程,早发型庞贝氏病的确诊只需要2小时,而且在4小时内就可以完成酶替代治疗的手续。早发型庞贝氏病儿童开始接受酶替代治疗的平均年龄是9天,位居全球首位。经他们治疗的患者有更好的生化生理指标、发育结果以及更少的免疫反应。独立行走的平均年龄为11个月,与正常儿童一样。因此,他们呼吁全世界应该普及庞贝氏病的新生儿筛查,因为早发现早治疗至关重要。
疾病介绍
1法布里病
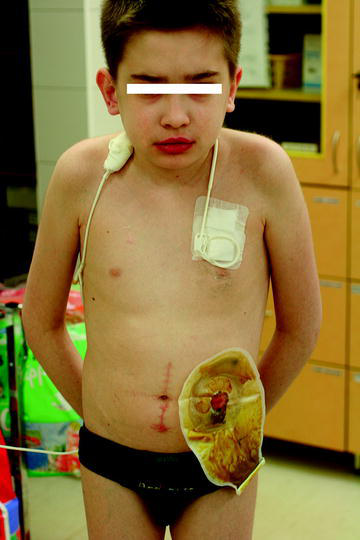 我怎么跟别的孩子不一样,为什么我不出汗,手指脚趾疼痛难忍?
我怎么跟别的孩子不一样,为什么我不出汗,手指脚趾疼痛难忍?
法布里病的临床危害
2 戈谢病
 为什么我的肚子那么大,还老是流鼻血
为什么我的肚子那么大,还老是流鼻血
戈谢病临床危害:
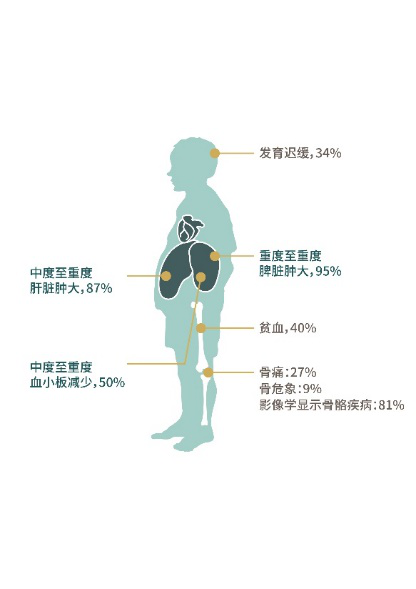
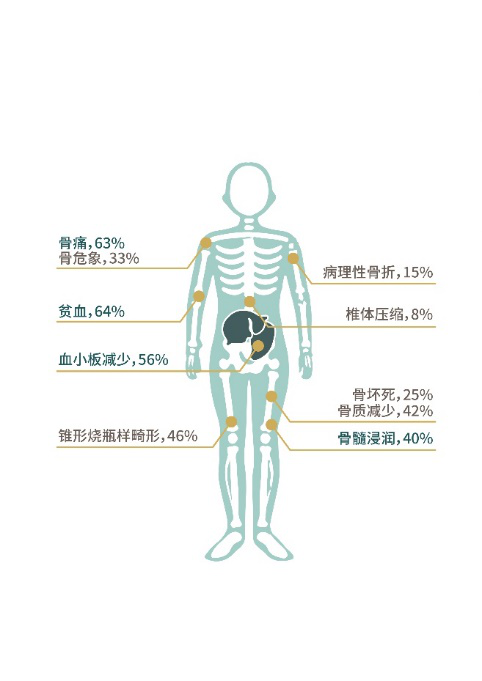
儿童患者的临床多脏器损害及发生率 成年患者的临床多脏器损害及发生率
3庞贝氏病
 我不想带呼吸机,我想出去和小朋友们一起玩
我不想带呼吸机,我想出去和小朋友们一起玩
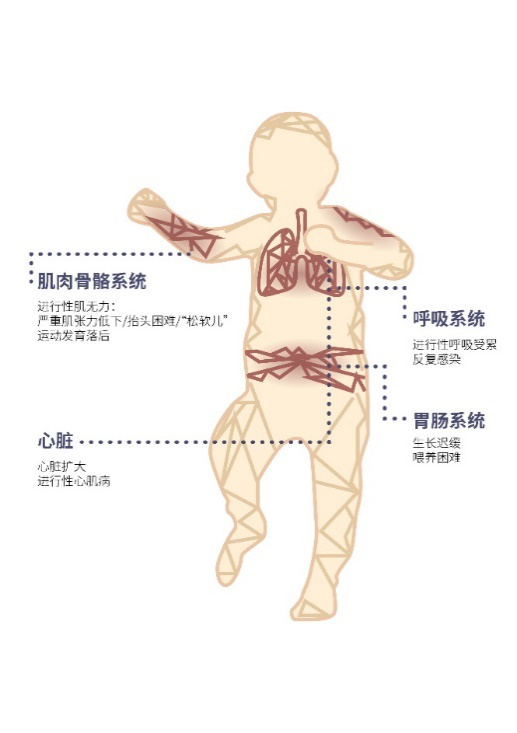
庞贝氏病的临床危害
婴儿型:研究显示,未接受治疗的婴儿型患儿
2个月龄(中位数)出现症状
5.9个月龄呼吸功能障碍,需呼吸肌支持
常于1岁左右死于心力衰竭及呼吸衰竭
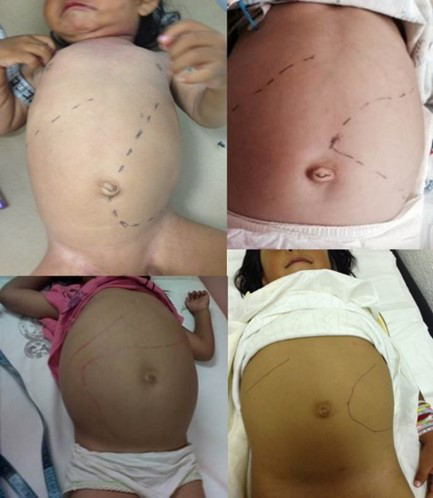
4 粘多糖贮积症I型
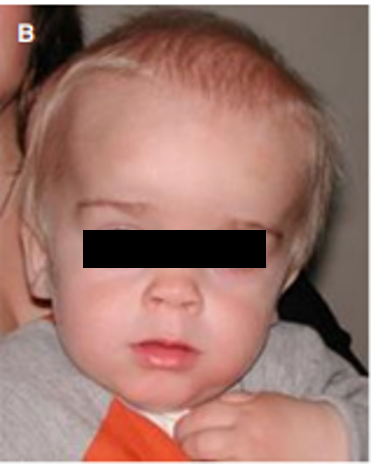 我好久都不长个了,小朋友们还笑话我长得难看,为什么我和他们不一样?
我好久都不长个了,小朋友们还笑话我长得难看,为什么我和他们不一样?
粘多糖贮积症I型临床危害

为什么要做新生儿筛查
早发现,早治疗,早干预
姐妹治疗开始时间的早晚,导致了治疗效果的巨大差异
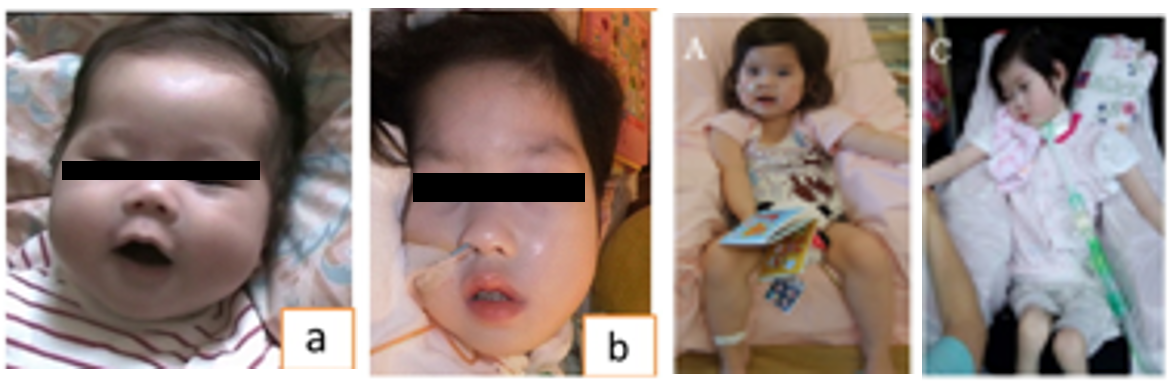
姐姐:先证者,4个月被诊断为庞贝氏病,随后开始酶替代治疗,治疗时已出现肌无力等症状。6岁的姐姐,卧床不起,需24小时带呼吸肌

妹妹:
出生10天确诊为庞贝氏病,12天开始进行酶替代治疗,治疗时未出现症状
3岁的妹妹正常发育,1岁时可独立行走,无心肺功能衰竭
治疗方法
酶替代治疗 异基因造血干细胞移植
戈谢病、庞贝氏病酶替代治疗药物思而赞、美而赞已在中国大陆上市,浙江、上海、青岛、宁夏、山西、湖南等省市已将这两种或一种特效药物纳入大病医保。
服务优势
提供筛-诊-治闭合体系的整体解决方案
精准筛查:结果判定简单,特异性及灵敏度高
快速确诊:筛查阳性的新生儿,进一步通过实验室自主研发的外周血白细胞酶活性检测结合基因检测快速确诊
协助治疗:一旦确诊,推荐合作专家,启动绿色通道,为患儿快速提供个体化治疗
筛查流程
1.知情同意
监护人签署《知情同意书》
2.标本采集
干血斑,3孔
3.报告周期
10个工作日出具检验报告
4.结果解读
筛查阴性:排除
筛查阳性:进一步进行外周血白细胞酶活性检测+基因检测确诊
常见问题
1 在孩子出生前我们已经做过很多检查,比如无创产前检测、唐氏筛查等等,还需要做这四种疾病的筛查吗?
答:目前这些产前筛查是无法检测出这四种疾病的,而且大多孩子在胎儿及出生时是表现正常的,仅从孩子的表现上是无法做到早发现早治疗的。因此,通过新生儿期的筛查可以快速发现患儿,从而给予及时治疗,避免或延缓患儿智力发育落后及残疾的发生。
2 我的孩子出生后看起来很正常,需要做筛查吗?
答:孩子一般在出生时大都表现正常,但由于体内缺乏某种溶酶体酶,会导致相应代谢物的不断累积,随着孩子的生长发育逐渐开始出现发育落后、智力低下等症状。一旦出现临床症状即使治疗大多不可逆,尤其是神经系统方面的损害。因此,在新生儿时期进行疾病筛查可以在症状前诊断,给予患儿及时治疗,可以极大的提高患儿的生活质量。
3 我们没有家族史,孩子有必要做筛查吗?
答:溶酶体病多为隐性遗传性疾病,也就是说基因致病突变携带者表现是正常的,当父母双方均为携带者时,孩子就会有一定的机率遗传到父母的致病突变,从而导致疾病的发生,另外孩子自发突变也会导致疾病。因此,没有家族史,并不代表孩子不会患病。
4 筛查的这几种疾病常见吗?
答:这几种疾病都属于罕见病,传统的文献报道发病率较低,但是,随着检测技术手段的提高及新生儿筛查的普及,其真实的发病率并不低,中国台湾对法布里病进行新生儿筛查后发现,男性婴儿的发病率达到1/1250。美国对法布里病、戈谢病、庞贝氏病、粘多糖贮积症I型的新生儿进行筛查,其总的发病率达到1/2000.
参考文献:
[1]Hopkins PV,et al. Incidence of 4 Lysosomal Storage Disorders From 4 Years of Newborn Screening[J]. JAMA Pediatr.2018.172(7):696-697.
[2] Roy W.A.Peake,et al. Newborn Screening for Lysosomal Storage Disorders [J]. J Pediatr Genet. 2017,6(1):51-60.
[2]Matsuoka T,et al. Divergent clinical outcomes of alpha-glucosidase enzyme replacement therapy in two siblings with infantile-onset Pompe disease treated in the symptomatic or pre-symptomatic state [J]. Mol Genet Metab Rep. 2016,9:98-105.
[3]Majed Dasouki,et al. Pompe Disease: Literature Review and Case Series [J]. Neurol Clin. 2014,32(3):751-ix
[4]Piotr Buda,et al. Gastrointestinal Phenotype of Fabry Disease in a Patient with Pseudoobstruction Syndrome [J]. JIMD Rep. 2012,4:25-28
[5] Rossella Parini,et al. International working group identifies need for newborn screening for mucopolysaccharidosis type I but states that existing hurdles must be overcome [J]. Acta Paediatr. 2018,107(12):2059-2065.
[6] Magdalena CR,et al. Improvement of life quality measured by Lansky Score after enzymatic replacement therapy in children with Gaucher disease type 1 [J]. Mol Genet Genomic Med. 2018,6(1):27-34
[7]中华医学会儿科学分会内分泌遗传代谢组.糖原贮积病II型诊断及治疗专家共识(2013).中华医学杂志.2013,93:1370-1373.
[8] 中国法布里病专家协作组.中国法布里病(Fabry病)专家共识(2013).中华医学杂志.2013,93(4):243-247.
[9] 中华医学会儿科学分会遗传代谢内分泌学组.中国戈谢病诊治专家共识(2015).中华儿科杂志.2015,53(4):256-261.
[10]罕见病诊疗指南,2019版