从苯丙酮尿症认识新生儿筛查
苯丙酮尿症是苯丙氨酸羟化酶活性缺陷导致苯丙氨酸代谢障碍的常染色体隐性遗传性疾病。
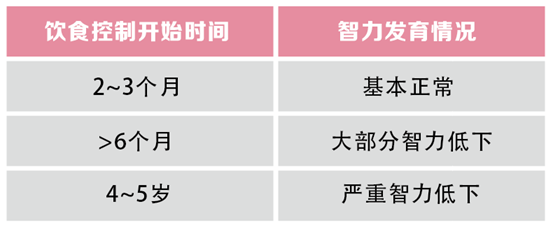
出生正常患儿如果吃母乳或含苯丙氨酸奶粉,因先天性苯丙氨酸羟化酶活性降低,不能充分代谢苯丙氨酸,使其体内不断蓄积,脱氨后生成过多苯丙酮酸,对孩子神经系统造成不可逆损伤。
新生儿期对苯丙氨酸羟化酶筛查异常并确诊疾病者,给予不含苯丙氨酸的特殊奶粉,可避免患儿出现神经系统的进行性损伤。
新生儿疾病筛查意义
⑴尽早发现携带有潜在致残或致命性疾病的新生儿,为早期干预提供时机;
⑵早期而有效地对因治疗,避免或减轻患儿重要器官的不可逆损害,提高患儿生存质量;
⑶践行三级预防理念,降低出生缺陷,提高人口素质。
溶酶体贮积症及相关四种酶的新生儿筛查
溶酶体贮积症是溶酶体内某种水解酶缺陷导致生物大分子物质不能降解而在溶酶体中堆积,使得溶酶体发生肿胀,细胞功能受损,而出现严重临床表现的一类疾病。该病总发病率为1/7000~1/8000,常见的包括戈谢病、法布里病、庞贝氏病及粘多糖贮积症I型。
新生儿酶学四项筛查疾病介绍
01
戈谢病
是由β-葡糖脑苷脂酶活性缺陷导致的疾病。主要症状为肝脾肿大、血细胞减少、凝血障碍、骨病,伴或不伴神经系统症状。急性神经系统受累患者病情进展较快,病死率高,一般2~4岁前死亡。
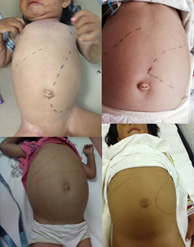
图:戈谢病患者表现为肝脾肿大
02
法布里病
是由α-半乳糖苷酶A活性缺陷导致的疾病。典型男性患儿多在儿童晚期或青少年早期以神经痛为首发症状,随后可出现角膜涡旋、血管角质瘤、胃肠疾病、蛋白尿等症状,疾病后期进展至肾衰竭、心脏及脑血管损害等。
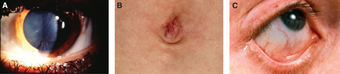
图:法布里病表现:A.角膜涡旋 B.血管角质瘤 C 眼球结膜血管迂曲
03
庞贝氏病
是由α-葡萄糖苷酶活性缺陷导致的疾病。临床分为婴儿型和晚发型。典型婴儿型主要表现为喂养困难、运动发育迟缓、松软儿、心脏扩大、肝脏肿大及舌体肥厚,疾病进展迅速,常于1岁左右死于心力衰竭及呼吸衰竭。
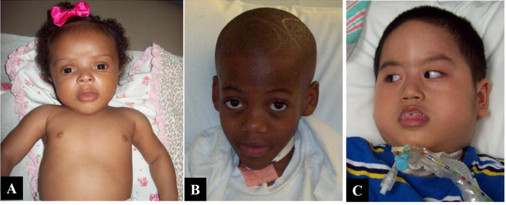
图:婴儿型庞贝氏病临床表现:呼吸困难
04
粘多糖贮积症I(MPS I)型
是由α-L-艾杜糖苷酸酶活性缺陷导致的疾病。重型患者临床表现为生长发育落后,面容粗陋,角膜混浊、青光眼,骨骼畸形、关节僵硬屈曲,腹部膨隆、肝脾肿大,呼吸道感染以及皮肤增厚、粗糙,耳聋,中耳炎等症状,不经治疗常10岁前死亡。
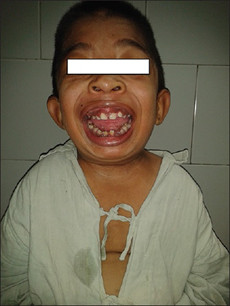
图:粘多糖贮积症I型患儿表现:面容特殊
为什么选择酶学四项进行筛查?
⑴ 有较好的治疗方法,如酶替代治疗药物,异基因造血干细胞移植等治疗可有效改善患者症状;早发现早治疗,显著改善预后,且开始治疗时间越早,效果越好;
⑵ 中国台湾已经把包括四项在内的多项溶酶体贮积症列入新生儿筛查项目;美国FDA 2017年批准了专门针对这四项的新生儿筛查系统;意大利、日本、德国、法国、澳大利亚等也均有开展相应疾病的筛查;
⑶ 2018年5月,五部委联合发布的121种罕见病目录,四项溶酶体贮积症收录其中;
⑷ 各省份已逐渐开始将四项相应疾病纳入医保,如浙江省已将戈谢病纳入医保、山西将戈谢病、庞贝氏病纳入医保等。
优势
提供筛-诊-治闭合体系的整体解决方案
精准筛查
针对疾病相对应的酶直接进行活性检测,对蛋白功能直接评价,不受年龄、状态、饮食等因素影响,结果判定简单,特异性及灵敏度高,假阴性率低;
快速确诊
筛查结果阳性的新生儿,应用外周血白细胞酶活性检测的自主研发专利技术,结合基因检测进一步快速确诊;
协助治疗
一旦确诊,立即帮助联系合作的北京三甲医院罕见病专家,启动专家绿色通道为患儿提供个体化治疗方案,并协助申请罕见病防治发展专项基金给予资金支持。
请问我的孩子很健康,需要做筛查吗?
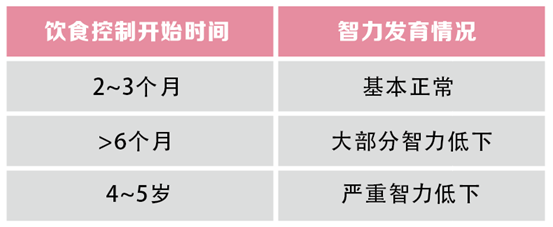
孩子一般在出生时大都表现正常,但由于体内缺乏某种溶酶体酶,会导致相应代谢物的不断累积,随着孩子的生长发育逐渐开始出现生长发育落后、智力低下等症状。一旦出现临床症状即使治疗大多不可逆,尤其是神经系统方面的损害。因此,在新生儿时期进行疾病筛查可以在症状前诊断,给予患儿及时治疗,可以极大的提高患儿的生活质量。
我们没有家族史,孩子有必要做筛查吗?
溶酶体病多为隐性遗传性疾病,也就是说基因致病突变携带者表现是正常的,当父母均为携带者时,孩子就会有一定的机率遗传到父母的致病突变,从而导致疾病的发生,另外孩子自发突变也会导致疾病。因此,没有家族史,并不代表孩子不会患病。