戈谢病(Gaucher disease)
时间:2020年07月22日信息来源:本站原创点击:次
戈谢病是一种常见的溶酶体病,为常染色体隐性遗传,属于罕见病范畴。人群中戈谢病的发病率接近1/60000 ~ 1/40000,在德裔犹太人群体中可达1/800,国内尚无确切的流行病学统计资料。
戈谢病临床表现
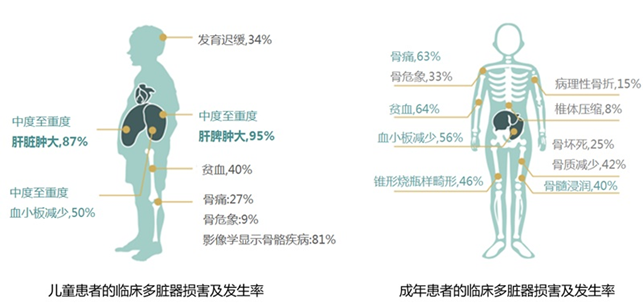
根据神经系统是否受累,主要分为3个亚型:慢性非神经病变型(I型)、急性神经型(II型)、亚急性神经型(III型),儿童与成年发病临床表现略有差异。
戈谢病发病机制:
GBA基因突变导致溶酶体内β-葡糖脑苷脂酶活性缺乏,其底物葡萄糖脑苷脂在肝、脾、骨骼、肺,甚至脑的巨噬细胞溶酶体中贮积,形成典型的“戈谢细胞”,导致受累组织器官出现病变。
主要诊断方法
酶学检测:外周血白细胞β-葡糖脑苷脂酶活性检测,当酶活性降低至正常值的30%以下时,可确诊。酶学检测是戈谢病诊断最有效,最可靠的方法。
GBA基因突变分析:确定致病变异位点,为临床遗传咨询和优生优育提供依据。
辅助诊断:骨髓形态学检查,血浆壳三糖酶活性检测等。
戈谢病治疗预后
酶替代治疗:药物Cerezyme(imiglucerase,伊米苷酶)。
造血干细胞移植
对症支持治疗:输血、骨科处理及脾切除等治疗。
下一篇:庞贝氏病(糖原贮积症II型)